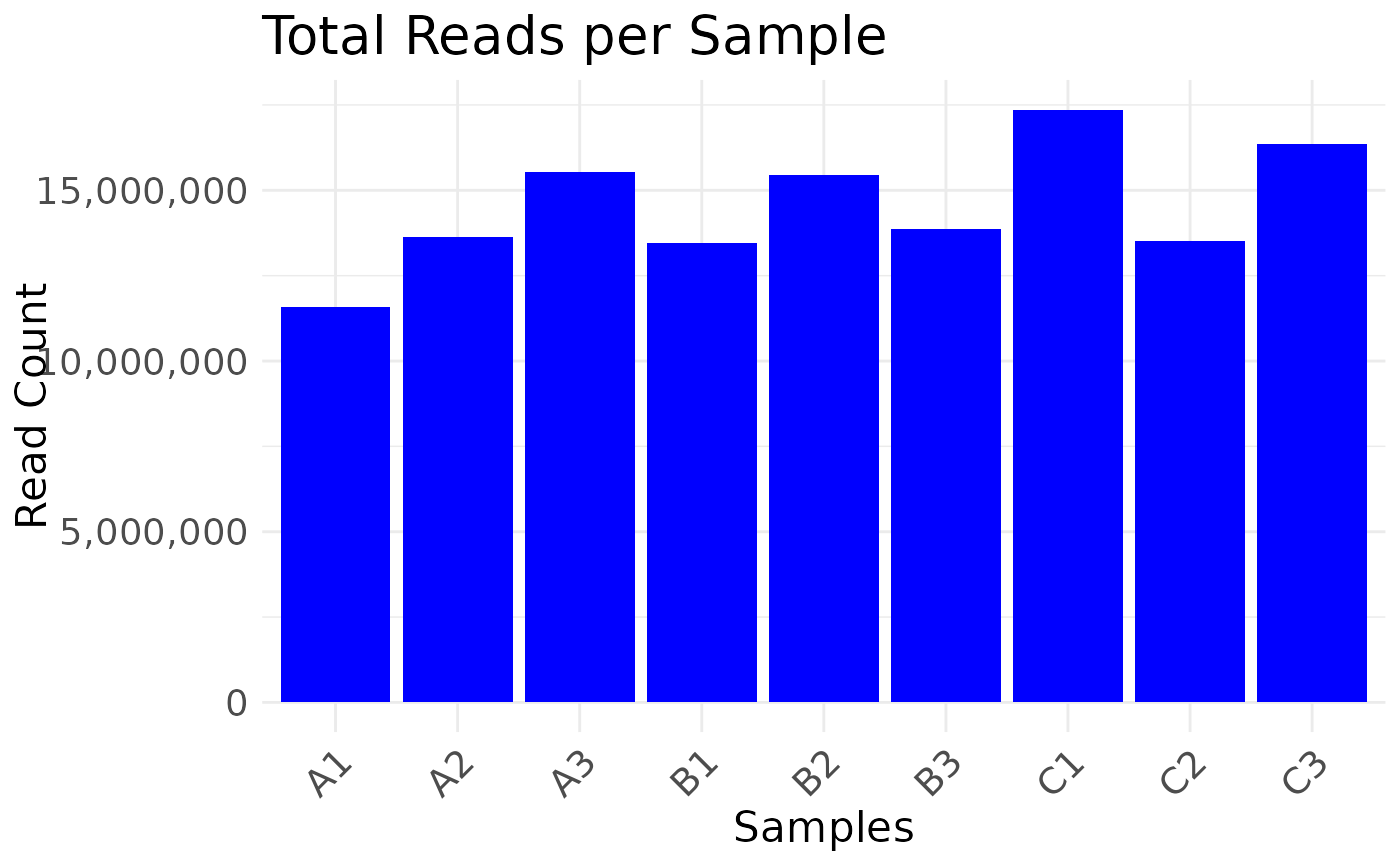
The first argument can be a multiOmicDataset object (moo) or a data.frame containing counts.
For a moo, choose which counts slot to use with count_type & (optionally) sub_count_type.
See also
Other plotters:
plot_corr_heatmap(),
plot_expr_heatmap(),
plot_histogram(),
plot_pca(),
print_or_save_plot()
Other moo methods:
batch_correct_counts(),
clean_raw_counts(),
diff_counts(),
filter_counts(),
filter_diff(),
normalize_counts(),
plot_corr_heatmap(),
plot_expr_heatmap(),
plot_histogram(),
plot_pca(),
run_deseq2(),
set_color_pal()
Examples
# multiOmicDataSet
moo <- multiOmicDataSet(
sample_metadata = nidap_sample_metadata,
anno_dat = data.frame(),
counts_lst = list(
"raw" = nidap_raw_counts,
"clean" = nidap_clean_raw_counts
)
)
plot_read_depth(moo, count_type = "clean")
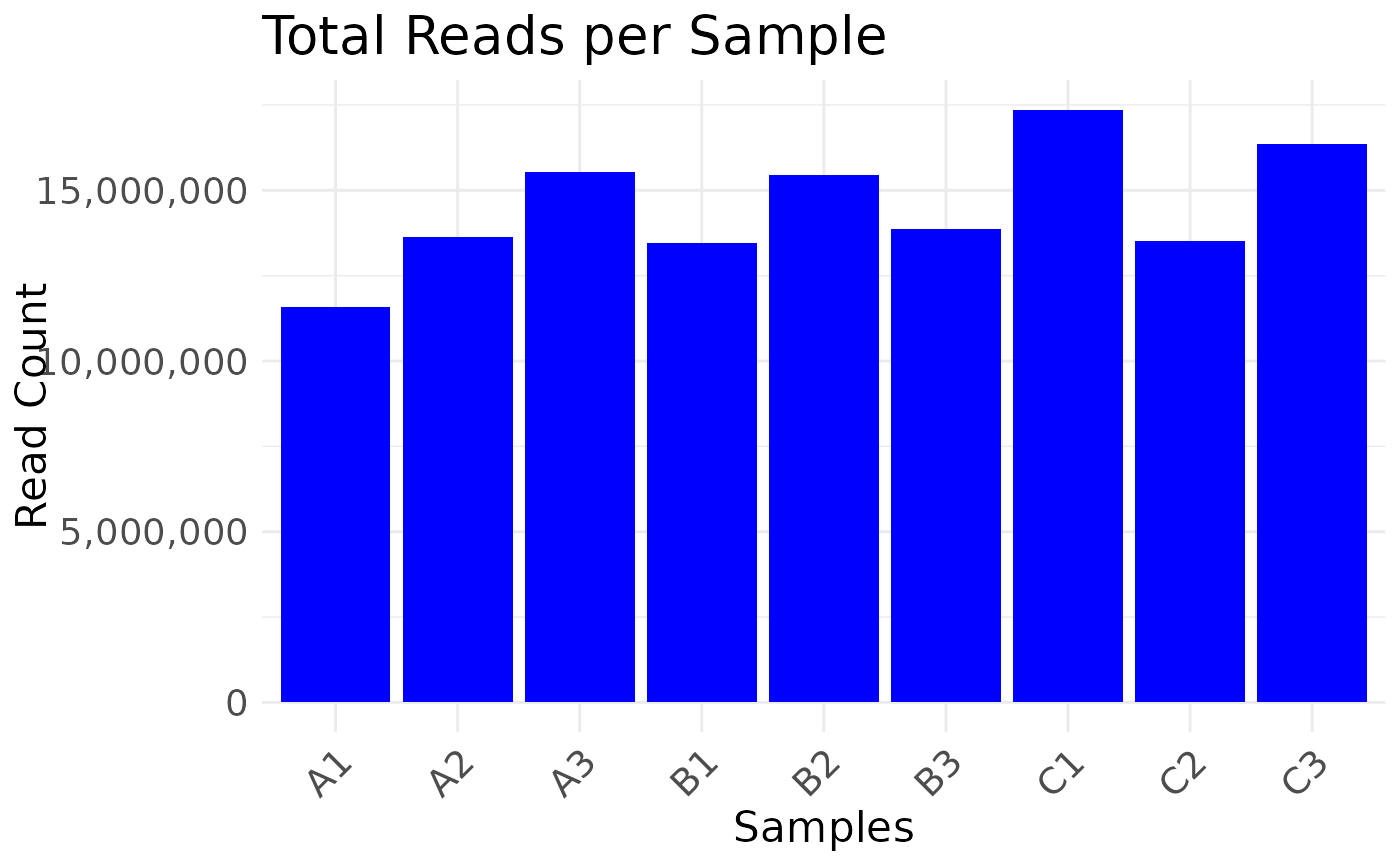 # dataframe
plot_read_depth(nidap_clean_raw_counts)
# dataframe
plot_read_depth(nidap_clean_raw_counts)